| CAS NO: | 1802929-43-6 |
| 规格: | ≥98% |
| 包装 | 价格(元) |
| 2mg | 电议 |
| 5mg | 电议 |
| 10mg | 电议 |
| 25mg | 电议 |
| 50mg | 电议 |
| 100mg | 电议 |
| Molecular Weight (MW) | 561.46 |
|---|---|
| Formula | C26H30Cl2N6O4 |
| CAS No. | 1802929-43-6 |
| Storage | -20℃ for 3 years in powder form |
| -80℃ for 2 years in solvent | |
| Solubility (In vitro) | DMSO: 100 mg/mL (178.1 mM) |
| Water: <1 mg/mL | |
| Ethanol: 6 mg/mL (10.68 mM) | |
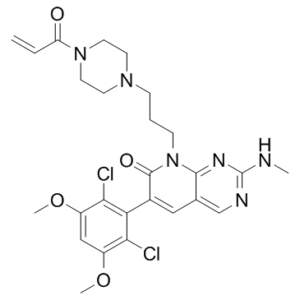
| SMILES Code | O=C1C(C2=C(Cl)C(OC)=CC(OC)=C2Cl)=CC3=CN=C(NC)N=C3N1CCCN4CCN(C(C=C)=O)CC4 |
| Synonyms | PRN-1371; PRN 1371; PRN1371; Chemical Name: 8-(3-(4-acryloylpiperazin-1-yl)propyl)-6-(2,6-dichloro-3,5-dimethoxyphenyl)-2-(methylamino)pyrido[2,3-d]pyrimidin-7(8H)-one Exact Mass: 560.1706 |
| In Vitro | In vitro activity: PRN1371 is an irreversible nanomolar inhibitor of FGFR1–4. PRN1371 presents a unique profile of high biochemical and cellular potency (FGFR1 IC50 = 0.6 nM, SNU16 IC50 = 2.6 nM), prolonged target engagement (FGFR1 occupancy 24 h = 96%),<30% 1='' herg='' inhibition='' at='' and='' good='' predicted='' adme='' stability='' with='' bme='' reactivity='' kd=''>100 μM. PRN1371 which maintained high FGFR1 occupancy with improved solubility and exceptional oral bioavailability. Kinase Assay: Enzyme inhibition is determined using a Caliper capillary electrophoresis system that separates phosphorylated and nonphosphorylated peptides on the basis of charge. Different concentrations of PRN1371 are first preincubated with enzyme for 15 min. The reaction is initiated with addition of peptide substrate, ATP, and Mg2+ and incubated at 25°C for 3 h. To stop the reaction, the mixture is quenched with EDTA. The buffer is 100 mM HEPES, pH 7.5, 0.1% BSA, 0.01% Triton X-100, 1 mM DTT, 10 mM MgCl2, 10 mM sodium orthovanadate, 10 μM β-glycerophosphate, and 1% DMSO. The ATP concentration of the reaction is at the predetermined value of the Km for ATP. Cell Assay: Human umbilical vein endothelial cells (HUVECs) were incubated in media supplemented with 10% FBS and seeded at 30 000 cells per well in a 96-well plate overnight. HUVECs were then transferred into serum free media 1 h before compound treatment. A compound concentration series was added to cells and incubated for 1 h at 37 °C. Cells were then stimulated with either 50 ng/mL of FGF2 or 50 ng/mL of VEGF for 10 min. Ice cold PBS was added to stop the reaction, and cells were washed three times to remove media. ERK phosphorylation is determined. |
|---|---|
| In Vivo | A rat iv (2 mg/kg) PK study of compound 34 showed rapid clearance (Cl = 160 ml/min/kg), yet dosing po (20 mg/kg) demonstrated high oral exposure (AUC = 4348 h·ng/mL) and a reasonable half-life (t1/2 = 3.8 h). PK studies of compound 34 in rat, dog, and cynomolgus monkey showed rapid iv clearance in all species; however there were large species differences in oral exposure and bioavailability for monkey compared to rat and dog. In rat, high exposure upon oral dosing (e.g., Cmax = 1785 ng/mL, AUC = 4348 ng·h/mL) and>100% bioavailability (F) suggested good absorption and partial saturation of clearance mechanisms at the 20 mg/kg dose. Unique to the rat, there is a large difference in half-life between the iv (t1/2 = 0.8 h) and po (t1/2 = 3.8 h) routes of administration, also indicative of possible saturation of a clearance mechanism upon oral dosing. In the dogs, the same methylcellulose suspension formulation used for the rat gave low oral absorption and bioavailability (F < 15%). In SNU16 gastric cancer xenograft mouse model, Compound 34 induced a dose-dependent reduction in tumor volume and up to 68% tumor growth inhibition at the highest dose of 10 mg/kg b.i.d. following 27 days of treatment. All doses were well tolerated with no significant body weight loss. |
| Animal model | SNU16 gastric cancer xenograft mouse model |
| Formulation & Dosage | 0.5% methylcellulose w/w in deionized water; 10 mg/kg; oral |
| References | J Med Chem. 2017 Aug 10;60(15):6516-6527. |
 m.cnreagent.com
m.cnreagent.com